Collection of Atherosclerosis research paper collection: A comprehensive systematic review paper on Lipoprotein Transport mechanisms, pathophysiology, and treatments
This research paper on atherosclerosis is a systematic review paper on the second stage of atherosclerosis, lipoprotein transport, its biological mechanisms, role in atherosclerosis, its connection to endothelial dysfunction, and exploring known treatments through accredited clinical and experimental studies.
RESEARCH
Avya Patel
4/12/202520 min read
Atherosclerosis research paper collection:
A comprehensive systematic review paper on Lipoprotein Transport mechanisms, pathophysiology, and treatments
Abstract on lipoprotein transport:
Background: Lipoprotein transport is the second hallmark sign of atherosclerosis, and comes as a result of endothelial dysfunction, more importantly, how the endothelium becomes increasingly permeable to LDL-cholesterol. Upon entering the endothelium, LDL-c goes under oxidation to become ox-LDL, later creating foam cells through macrophages. This creates a wide array of problems in the intima of the blood vessels. This pathway allows for oxidative stress, continuing atherosclerosis.
Objective: This review systematically evaluates the pathophysiological mechanisms underlying lipoprotein transport and explores known pharmacological interventions aimed at restoring endothelial function.
Methods: A systematic review of existing literature was conducted following PRISMA guidelines. Studies investigating endothelial dysfunction and its unique inhibition of mechanisms. PCSK9 Inhibitors, Bile Acid Sequestrants, and Ezetimibe were investigated in clinical and experimental studies with their accordance of biological mechanisms in endothelial dysfunction.
Results: Results confirm the effect of known treatments by further consolidating specific evidence of lowering LDL-c, upregulating LDLR and HDL-c, all allowing to suppress hypercholesterolemia. PCSK9, evolocumab is a key treatment by lowering LDL-c by 59%, PSCK9 inhibitors limit the production of Lp(a) while increasing HDL-c. Both Bile Acid Sequestrants, and Ezetimibe allow for an upregulation of LDL-receptors on liver cells to pull LDL-c out of the blood, however both treatments have their specific mechanism of action as discussed in this paper.
Conclusion: Lipoprotein transport serves as a critical target for lowering LDL-c in circulating blood. Current pharmacological therapies, PCSK9 Inhibitors, Bile Acid Sequestrants, and Ezetimibe are efficient treatments in lowering LDL-c and other “bad” lipoproteins to improve full body health, but limitations exist in long-term clinical outcomes. Further research can allow the integration of individualized therapy based on genetic, metabolic, or lipid profile factors, as well as RNA-based therapies, CETP inhibitors, HDL mimetics.
Introduction on Lipoprotein transport in Atherosclerosis:
Atherosclerosis, otherwise known as arteriosclerosis is defined as the buildup of plaque made out of fats, cholesterol and other substances in and on the artery walls [1]. This accumulation of plaque restricts blood flow (ischemia) over time which can lead to a myocardial infarction (heart attack). Additionally, this buildup of plaque can lead to a blood clot or thrombosis when ruptured. Atherosclerosis is not specific to only the heart, however subdivides into several other diseases including Coronary Artery Disease (CAD/ CHD) which is the most common form of atherosclerosis, in summary, atherosclerosis serves as an underlying cause of several more specific cardiovascular diseases. Although specific data on atherosclerosis is limited, the effect of atherosclerosis is seen in broader cardiovascular data collections. For instance, in 2022, heart disease remained the leading cause of death in the United States, accounting for 702,880 deaths. In this number, CHD (Coronary heart disease) caused 371,506 deaths in 2022, CHD remains the most common form of heart disease and atherosclerosis [2]. Around every 40 seconds, someone in the US experiences a myocardial infarction which stems from a form of atherosclerosis called CAD [2]. Atherosclerosis is divided into 4 major stages including endothelial dysfunction, lipoprotein accumulation, oxidative stress, and arterial calcification. [4], [5], [6], [7]. This research paper will focus solely on lipoprotein transport and its role in atherosclerosis, more specifically, how cholesterol accumulation exaggerates the effects of endothelial dysfunction in Atherosclerosis. The complex interplay of lipoprotein transport as well as discussion for treatments, inhibition and the potential for future studies will be discussed in this paper. Understanding the mechanisms attributed to each stage of atherosclerosis is necessary to pioneer a new treatment or inhibition method. A systematic review of lipoprotein transport and the role of cholesterol accumulation is necessary to evaluate known treatments for atherosclerosis and CAD in general by dividing atherosclerosis into 4 stages and assessing the most effective pharmaceutical treatments for atherosclerosis to develop a foundation where future treatments with the help of technology will come with ease. Finally, the objective of this systematic review is to analyze how modern pharmacological strategies target key mechanisms in lipoprotein transport, and eventually plaque formation progression.
Materials and Methods:
This study evaluated published clinical data from databases such as PubMed, Google Scholar, ClinicalTrials.gov to examine published peer-review studies and human clinical trials to assess the biomechanical effects of known treatments for lipoprotein transport. It also adhered to the standards outlined in the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) Statement [8].
3.1: Literature Search Strategy
A comprehensive search strategy was implemented to find accredited sources such as CDC, WHO, and other electronic databases such as PubMed, Google Scholar, ClinicalTrials.gov were used to isolate potential clinical trials and/ or experimental trials of a given pharmaceutical treatment. Free-text keywords related to "lipoprotein transport treatments,", “cholesterol in atherosclerosis”, "PCSK9 Inhibitors", "Bile Acid Sequestrants", and "Ezetimibe".
The mechanism of action of each treatment was also researched with its biological effect on lipoprotein transport.
3.2: Study Selection Criteria
The selection process was conducted in accordance with the PRISMA guidelines. Independent reviewers screened titles and abstracts to identify viability of articles in accordance with the. The full texts of the selected articles were independently evaluated by the same reviewer to determine their eligibility for inclusion in the meta-analysis or experimental research of a large sample size.
The inclusion criteria were as follows: Study types including; (1) Clinical trials (randomized controlled trials, cohort studies), (2) Systematic reviews & meta-analyses, (3) In-vitro or mechanistic studies with direct relevance to human atherosclerosis. Publication dating as followed; 2005–2025 (Ensure the study reflects current pharmaceutical strategies). Population specifics including; (4) Studies involving human participants diagnosed with atherosclerosis or at high cardiovascular risk, (5) Studies that explore age-related differences in treatment responses. Interventions which analyzed pharmaceutical interventions targeting lipoprotein transport (4.2)
The exclusion criteria were as follows: (1) Animal studies; Unless they provide direct translational relevance to human pathophysiology. (2) In-vitro studies without human application; Studies focusing purely on cell cultures or animal models without clear clinical implications. (3) Case Reports, Opinion Pieces, or Editorials. (4) Studies with small sample sizes (<100 participants), unless the study is incredibly novel and pinpoints the mechanism of action of the pharmaceutical treatment. Interventions including: (1) Surgical or interventional procedures (e.g., stenting, bypass surgery), (2) Alternative or herbal medicine studies without clinical validation and rigorous human clinical trials, (3) Studies published before 2005 and studies not in English.
3.3: Data Extraction & Analysis
A data extraction method was used to collect relevant information from selected papers and studies. An independent reviewer extracted data including relevance of the abstract, sample Size (Age, Gender, Comorbidities, Inclusion/Exclusion Criteria), treatment used (Drug Name, Dosage, Duration, Mechanism of Action), Atherosclerosis Stage Targeted (Endothelial Dysfunction, Lipoprotein Transport, Oxidative Stress, Calcification), outcome measures (Primary ones including LDL-C Reduction, Inflammatory Markers (CRP, IL-1β), Arterial Plaque Reduction and secondary ones including Cardiovascular Events, Adverse Effects, Mortality Reduction). This data was put through a PRISMA flowchart [8] for study selection and it was synthesized to identify patterns, trends, and gaps in the current literature. The studies were assessed through qualitative (comparisons in treatment effectiveness) and quantitative measures (treatment trend and analysis).
Pathophysiology of Lipoprotein transport & Targeted Treatments
4.2 Lipoprotein transport & role of cholesterol (LDL) in plaque formation
After the emergence of endothelial dysfunction, lipoprotein transport leads to LDL (low density lipoprotein) cholesterol to be oxidized, leading to foam cell formation. Here, foam cells are essentially macrophages that engulf cholesterol deposits. Recently, the emergence of LP-a (lipoprotein a) has been noted as a risk factor for atherosclerosis and CAD. Lp(a) has many functions, which include proatherosclerotic, prothrombotic and pro-inflammatory roles [11]. Before getting into the specific pathophysiology of lipoprotein transport, it is essential to understand how the movement of LDL cholesterol and other triglycerides contribute to atherosclerotic plaque formation. Normally, lipoprotein transport occurs when LP (lipoproteins) transport fats, as mentioned above, through the blood stream. However, in atherosclerosis, due to endothelial dysfunction, lipoprotein transport is affected, leading to plaque formation via higher levels of LDL circulation. In the arterial walls, lipoproteins can be modified by oxidation, forming oxLDL [12]. oxLDL are more likely to be engulfed by macrophages, leading to a higher chance of foam cell formation. This be known, the specific pathophysiology attributed to lipoprotein transport will be discussed below.
Pathophysiology of Lipoprotein Transport:
The primary pathophysiologic effects attributed to lipoprotein transport in atherosclerosis are as followed, oxidative LDL formation, followed by foam cell production, how foam cells lead to inflammation and plaque growth, HDL undoes the plaque formation by LDL by reverse cholesterol transport [13].
Foam cell formation via formation of oxLDL species:
In a brief summary, Low-density lipoprotein (LDL) cholesterol penetrates the arterial wall due to endothelial dysfunction and undergoes oxidation. This oxidized LDL is then engulfed by macrophages, leading to the formation of foam cells.
How LDL undergoes oxidation to form oxLDL (Oxidative Modification Hypothesis) [14]:
The oxidation of LDL was considered the main atherogenic modification of LDL within the vascular wall for decades. The Oxidative Modification Hypothesis is a theory proposed which states that after LDL-c (LDL cholesterol) enters the arterial wall’s endothelium due to endothelial dysfunction, it undergoes multiple modifications that change their size, density, and chemical properties within the blood flow and vascular wall in which oxidation is the last stage in this cascade resulting in the atherogenic properties. The main factor underlying oxidative stress is a disbalance between radical production (reactive oxygen and/or nitrogen species formation) and radical scavenging systems (the antioxidant defense system).
OxLDL comes from metal ions, ROS, and major enzymes mediated modifications such as oxidase, lipase, myeloperoxidase. However, the gradual oxidation of LDL can be observed after LDL is trapped by the proteoglycans in the extracellular matrix of the endothelium. As mentioned above, oxLDL goes under both enzymatic and non-enzymatic alterations, such are, Desialylation Aggregation, Enzymatic degradation (via lipases and proteases). Given endothelial dysfunction, ROS are produced by enzymes such as NADPH oxidase, Myeloperoxidase, and Xanthine oxidase. In the LDL’s phospholipid shell, there is a presence of polyunsaturated fatty acids (PUFAs) which are oxidized by the reactive oxidative species, these conversions to the phospholipid shell finally allow LDL to be converted into oxLDL.
OxLDL stimulates endothelial cells by the activation of adhesion molecules, these adhesion molecules allow blood-leukocytes to bind to endothelial cells and later, migrate into the intima of a blood vessel (inner portion of the blood vessels past the smooth muscle layer). Such adhesion molecules allow for the migration of macrophages to the site of oxLDL. Finally, oxLDL is recognized by scavenger receptors such as CD36 and SR-A, which then promotes inflammatory cytokines, allowing for macrophage migration.
Engulfing formed oxLDL species creates foam cells from macrophages [15]:
Monocytes are differentiated into macrophages which start to take in excess lipids in the arterial sub-endothelial space. A possible explanation of macrophage migration to the arteries lies in the release of inflammatory cytokines such as tumor necrosis factor-alpha due to endothelial dysfunction. When macrophages take up excess lipids, they collect in the cytoplasm as cytoplasmic lipid droplets, such induced swelling leads macrophages to be transformed into foam cells (characterized by their foamy appearance and lipid-filled vacuoles). The accumulation of foam cells in atherosclerosis lead to fatty streaks and plaque developments, induce inflammation, and contribute to plaque growth. When such foam cells rupture, it releases its lipid contents which can lead to a larger plaque deposit or thrombosis.
Foam cells trigger inflammation, continuing plaque growth [16], [17]:
The excessive uptake of oxLDL not only converts these macrophages into foam cells but also exerts changes on intracellular signaling pathways in a manner that favors proinflammatory responses, thereby conferring proinflammatory traits on foam cells as the plaque advances [16]. The recruitment of other lymphocytes to areas of plaque occur due to pro-inflammatory signalling pathways in foam cells. Another explanation as to how foam cells continue plaque growth lies outside of inflammation, instead, points to necrosis due to oxLDL where lipid peroxides modify cellular proteins and lipids, trigger oxidative stress, and modulate numerous signaling pathways and the expression of diverse genes, essentially resulting in the formation and deposition of cellular debris as apoptotic and necrotic bodies. [16] Explains the role of hypoxia and how upregulation of the HIF-1α transcription factor in plaque macrophages leads to the transactivation of proinflammatory genes such as cytokines.
The continuation of plaque growth by foam cells is observed in [18] and [19]. Such continuation is specifically understood in terms of lipid accumulation and its role in the various apoptosis cascades below.
The cytochrome c, a mitochondrial intermembrane protein, is delivered into the cytosol and induces the expression of apoptotic protease activating factor 1 (APAF1) and apoptosome, which triggers apoptosis cascade by activating the serine protease caspase 9. Active caspase 9 stimulates caspase 3 and caspase 7, which results in DNA fragmentation [19].
The stimulation of the NF-κB pathway and the NOD-like receptor 3 (NLRP3) inflammasome causes Pyroptosis which is a highly inflammatory form of programmed necrosis which is initially identified in infections of bacterial and other pathogens [17]. Pyroptosis also promotes necrotic core formation in advanced atherosclerosis.
HDL removes excess cholesterol via reverse cholesterol transport [20]:
The ability of HDL to perform reverse cholesterol transport (RCT) is a novel study, leading to speculations of how HDL can be used for atheroprotection.
HDL plays a crucial protective role in cardiovascular health by mediating RCT, a process that removes excess cholesterol from peripheral tissues and atherosclerotic plaques. HDL particles collect cholesterol from macrophages and foam cells in the arterial wall and transport it to the liver for excretion in bile. This process begins with HDL interacting with ATP-binding cassette transporters like ABCA1 and ABCG1 on the surface of macrophages, facilitating the efflux of cholesterol onto nascent HDL particles. Additionally, when the free cholesterol esterified in HDL becomes very hydrophobic, it is pushed to the core of the lipoprotein, away from contact with the water medium. The efflux to HDL involves passive diffusion of cholesterol as well as active cholesterol transfer. The enzyme LCAT (lecithin-cholesterol acyltransferase) then esterifies the free cholesterol, allowing it to be sequestered into the core of mature HDL particles. Ultimately, HDL delivers this cholesterol to the liver via scavenger receptor class B type 1 (SR-BI), where it is eliminated from the body.
Targeted treatments for Lipoprotein Transport:
Known treatments for Lipoprotein transport are PCSK9 Inhibitors such as Evolocumab and Alirocumab (increase LDL receptor recycling), Bile Acid Sequestrants such as Cholestyramine to make the liver use cholesterol for bile production, and Ezetimibe to reduce intestinal cholesterol reabsorption (this treatment works increasingly well with Statin therapies for endothelial dysfunction).
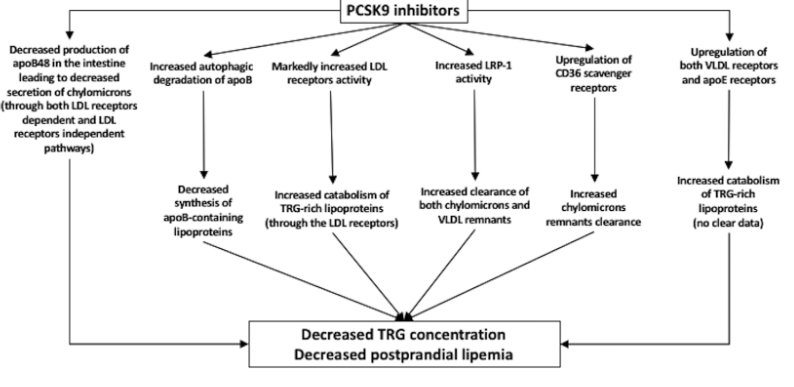
PCSK9 Inhibitors and lipoprotein transport [11], [21]:
PCSK9 or Proprotein convertase subtilisin/kexin type 9 inhibitors are emerging lipid-lowering agents that can effectively reduce Lp(a) levels.
Several studies such as the FOURIER trial indicate significant reduction of TRG (Triglycerides) [22]. Out of a randomized study of 7,564 patients with atherosclerotic cardiovascular disease and LDL cholesterol levels of 70 mg per deciliter (1.8 mmol per liter), this study expressed how the PCSK9 inhibitor evolocumab lowered LDL cholesterol levels by 59% from baseline levels as compared with placebo, from a median of 92 mg per deciliter (2.4 mmol per liter) to 30 mg per deciliter (0.78 mmol per liter). Lp(a) is a category of LDL-c, more thoroughly expressed in [21]. Although similar to LDL-c, Lp(a) has an apolipoprotein (a) group that is thought to make it more "sticky" and prone to clumping in blood vessels. Several studies have shown the decrease of Lp(a) levels after treatment of PCSK9 inhibitors. Results include a reduction of nearly 29% in a sample size of 4915, and a significant decrease of 48.6% of Lp(a) levels in a sample size of 812 when treatment groups were given 420mg/4 weeks duration. This data was collected by [21]. Finally, [21] shows the effect PCSK9 inhibitors have in increasing HDL-c, therefore, allowing for reverse cholesterol transport. in the Long-term Safety and Tolerability of Alirocumab Versus Placebo on Top of Lipid-Modifying Therapy in High Cardiovascular Risk Patients With Hypercholesterolemia (ODYSSEY Long Term) study, alirocumab administration (150 mg/2 weeks) was associated with an increase in HDL-C and apoA1 concentration by 4.6% and 2.9% [21].
Bile Acid Sequestrants and lipoprotein transport:
Bile acid sequestrants are pharmacologic molecules that bind to bile acids in the intestine resulting in the interruption of bile acid homeostasis and, consequently, reduction in low-density lipoprotein cholesterol levels in hypercholesterolemia [24]. This leads to increased fecal excretion of cholesterol and subsequent effects on lipoprotein transport. In a clinical trial which examined the role of colestilan as a Bile Acid Sequestrant, patients saw a decrease of LDL-C levels by 22.5% in a sample size of 183, colestilan also increased HDL-C by 6.6%, this study examined hypercholesterolemia (LDL-C, >160 mg/dL). An additional meta-analysis study [26] showed how cholestyramine as a Bile Acid Sequestrant in men with hypercholesterolemia resulted in overall reductions in total cholesterol and low-density lipoprotein cholesterol (LDLC) of 13.4% and 20.3%, respectively, compared with placebo (4.9% and 7.7%, respectively), suggesting the excretion influenced by cholestyramine.
Ezetimibe and lipoprotein transport:
In comparison to Bile Acid Sequestrants (e.g., cholestyramine, colestilan), Ezetimibe targets the small intestine, more specifically, the Niemann-Pick C1-Like 1 (NPC1L1) transporter in the small intestine. It is important to note that Ezetimibe is combined with statin therapies to further enhance the lowering of LDL-C. [27] Examined a combination therapy for Lipoprotein Transport with Ezetimibe and simvastatin. Out of 325 patients, the change from baseline in the mean (±SE) intima–media thickness of the carotid artery, was 0.0058±0.0037 mm in the simvastatin-only group and 0.0111±0.0038 mm in the combined-therapy group. This suggests how a combination therapy allows for a larger reduction in the intima-media thickness of the carotid artery. A final examination of Ezetimibe as a therapy for lipoprotein transport [28] indicates changes in LDL cholesterol −10.6%, in total cholesterol −9.53% in VLDL cholesterol −9.83% were detected significantly (P<0.01) (P<0.05). Later results also showed how a combination therapy of atorvastatin + Ezetimibe allowed for significant reduction in LDL-C while upregulating HDL-C.
Discussion of Lipoprotein Transport:
Lipoprotein transport is the second stage of Atherosclerosis, coming after endothelial dysfunction. These two phases go together as endothelial dysfunction allows the introduction of LDL lipoproteins to enter the intima and endothelium of blood vessels, allowing for the development of foam cells through oxidation of LDL cholesterol.
LDL-C is low density lipoprotein, and creates fatty deposits in the arterial walls, contributing to macrophage phagocytosis and the formation of an atherosclerotic plaque. Contrasting this, HDL-C is high density lipoprotein, and in the cascade of pathophysiology of lipoprotein transport, HDL-C allows for reverse cholesterol transport, slowing plaque buildup.
This paper delved into the specific pathophysiology attributed to lipoprotein transport. After entering the endothelium matrix, LDL-C undergoes oxidation through enzymatic conditions and non-enzymatic pathways. This oxLDL, due to endothelial dysfunction’s release of inflammatory markers, attracts macrophages, after engulfing oxLDL, macrophages turn to foam cells, characterized by extreme lipid deposits in these cells, which can lead to necrosis and apoptosis. These foam cells send out inflammatory markers, which in turn lead to higher amounts of macrophage migration, creating an early hallmark of atherosclerosis. However, with lipoprotein transport comes reverse cholesterol transport with the help of HDL-C, this process begins with HDL interacting with ATP-binding cassette transporters like ABCA1 and ABCG1 on the surface of macrophages, facilitating the efflux of cholesterol onto nascent HDL particles.
Additionally, this paper looked at key treatments for lipoprotein transport including PCSK9/Proprotein convertase subtilisin/kexin type 9 Inhibitors such as Evolocumab and Alirocumab (increase LDL receptor recycling), Bile Acid Sequestrants such as Cholestyramine to make the liver use cholesterol for bile production as well as targets the intestine’s ileum, and Ezetimibe to reduce intestinal cholesterol reabsorption (this treatment works increasingly well with Statin therapies for endothelial dysfunction).
In terms of PCSK9/Proprotein convertase subtilisin/kexin type 9 inhibitors and lipoprotein transport, this paper first investigated evolocumab as a possible treatment. In clinical studies, evolocumab lowered LDL cholesterol levels by 59% from baseline levels as compared with placebo, from a median of 92 mg per deciliter (2.4 mmol per liter) to 30 mg per deciliter (0.78 mmol per liter). This suggests how the inhibition of the role that PCSK9 play, which is binding to LDL receptors and marks them for destruction inside the liver cell, therefore, shows how drugs such as evololcumab is a monoclonal antibody that binds to PCSK9, stopping it from destroying LDL receptors. This means more LDL receptors survive and recycle, ensuring lower levels of oxLDL in the circulating bloodstream, therefore inhibiting atherosclerotic plaque development. PSCK9 inhibitors in this paper also are shown to lower Lp(a) (similar to LDL, but with an apolipoprotein (a) group that is thought to make it more "sticky" and prone to clumping in blood vessels. Results include a reduction of nearly 29% in a sample size of 4915, and a significant decrease of 48.6% of Lp(a) levels in a sample size of 812 when treatment groups were given 420mg/4 weeks duration. This suggests how PSCK9 inhibitors disrupt the formation of Lp(a) by not allowing the attachment of an apolipoprotein (a) group. Finally, the role of PSCK9 inhibitors and its upregulation of HDL-C is widely unknown, however this paper showed that the Hypercholesterolemia (ODYSSEY Long Term) study, alirocumab administration (150 mg/2 weeks) was associated with an increase in HDL-C and apoA1 concentration by 4.6% and 2.9%. This LDL receptors-mediated increased catabolism of Lp(a) was also confirmed by a recently published study, which showed that alirocumab was followed by a decreased Lp(a) concentration due to increased apo(a) clearance. [21]
Next, this paper pooled together data regarding Bile Acid Sequestrants (cholestyramine and colestilan and lipoprotein transport). Colestilan patients saw a decrease of LDL-C levels by 22.5% in a sample size of 183, colestilan also increased HDL-C by 6.6%, this study examined hypercholesterolemia (LDL-C, >160 mg/dL). Cholestyramine patients saw overall reductions in total cholesterol and low-density lipoprotein cholesterol (LDLC) of 13.4% and 20.3%. These two Bile Acid Sequestrants have similar roles in terms of binding bile acids and prevent their reabsorption, leading to increased excretion in the feces. In response, the liver uses cholesterol to synthesize new bile acids, which depletes its cholesterol stores from both the blood and tissues. The liver upregulates LDL receptors, pulling more LDL cholesterol from the bloodstream; this mechanism of action is seen above with PSCK9 inhibitors. This enhanced clearance of LDL particles reduces circulating LDL-C levels, making bile acid sequestrants effective in treating hyperlipidemia, especially when used in combination with other lipid-lowering therapies.
The investigation of Ezetimibe and lipoprotein transport was assessed as a specific lipoprotein transport treatment. It is crucial to understand that most studies use Ezetimibe with the combination of statin therapies in order to further enhance the lowering of LDL-C. Such studies were examined in this paper. One study used a combination therapy of ezetimibe and simivastatin, out of 325 patients, the change from baseline in the mean (±SE) intima–media thickness of the carotid artery, was 0.0058±0.0037 mm in the simvastatin-only group and 0.0111±0.0038 mm in the combined-therapy group. Furthermore, a study isolated Ezetimibe as one treatment, and indicated changes in LDL cholesterol −10.6%, in total cholesterol −9.53% in VLDL cholesterol −9.83% were detected significantly (P<0.01) (P<0.05). Knowing that Ezetimibe works specifically on the small intestine, and the Niemann-Pick C1-Like 1 (NPC1L1) transporter, it is safe to say that Ezetimibe is a valid treatment for lipoprotein transport by blocking NPC1L1 transporter, ezetimibe reduces the amount of cholesterol absorbed into the bloodstream. In response, the liver increases its expression of LDL receptors to extract more LDL cholesterol from the blood, thereby lowering circulating LDL-C levels and influencing overall lipoprotein transport in favor of reducing the risk of atherosclerotic cardiovascular disease.
Although Ezetimibe and Bile Acid Sequestrants have similar mechanisms by working on the intestine to upregulate LDL-receptors to lower LDL-C in the blood, they contrast each other in many ways. Ezetimibe inhibits NPC1L1 transporter to reduce intestinal absorption of cholesterol, whereas Bile Acid Sequestrants Binds bile acids, preventing their reabsorption to ensure the liver uses cholesterol to make more bile. Ezetimibe is commonly used with statins, whereas Bile Acid Sequestrants are often used solely to minimize GI tract complications.
For future research, a combinational therapy can be developed to inhibit endothelial dysfunction while also lowering circulating levels of LDL-C like Ezetimibe. It is important to note that with such targeted lipoprotein transport treatments, statins are often used to lower circulating cholesterol as mentioned in Collection of Atherosclerosis Research Papers: Endothelial Dysfunction. Future directions point to the integration of individualized therapy based on genetic, metabolic, or lipid profile factors, as well as RNA-based therapies, CETP inhibitors, HDL mimetics.
Conclusion on Lipoprotein transport:
Lipoprotein transport plays a key role in the development of Atherosclerosis for many reasons. Firstly, foam cell production is a hallmark sign of Atherosclerosis. After endothelial dysfunction, lipoprotein transport follows, starting with the formation of oxLDL species due to free radical oxidative species. Macrophages go to the injured endothelium and engulf on the oxLDL, creating foam cells, which increase the size of plaques, create inflammatory signals, and when apoptosis occurs, these foam cells release cytokines which lead to excess lymphocyte migration, all contributing to a larger plaque formation.
Current and noted therapies noted in this paper are PCSK9 Inhibitors such as Evolocumab and Alirocumab (increase LDL receptor recycling), Bile Acid Sequestrants such as Cholestyramine to make the liver use cholesterol for bile production, and Ezetimibe to reduce intestinal cholesterol reabsorption (this treatment works increasingly well with Statin therapies for endothelial dysfunction).
Later emphasis can be put upon creating multi-target treatment strategies with the integration of technological developments for personalized medicine to optimize lipid management and reduce cardiovascular risk.
Recognizing endothelial dysfunction and lipoprotein transport as a key biomarker of cardiovascular disease risk may shift clinical practice toward the early stages of atherosclerosis without the need of later surgical interventions.
References:
Mayo Clinic. (2024, September 20). Arteriosclerosis / atherosclerosis - symptoms and causes. Mayo Clinic; Mayo Clinic Staff. https://www.mayoclinic.org/diseases-conditions/arteriosclerosis-atherosclerosis/symptoms-causes/syc-20350569
CDC. (2024, May 20). Heart Disease Facts. Heart Disease. https://www.cdc.gov/heart-disease/data-research/facts-stats/
El Hadri, K., Smith, R., Duplus, E., & El Amri, C. (2021). Inflammation, Oxidative Stress, Senescence in Atherosclerosis: Thioredoxine-1 as an Emerging Therapeutic Target. International Journal of Molecular Sciences, 23(1), 77. https://doi.org/10.3390/ijms23010077
Sitia, S., Tomasoni, L., Atzeni, F., Ambrosio, G., Cordiano, C., Catapano, A., Tramontana, S., Perticone, F., Naccarato, P., Camici, P., Picano, E., Cortigiani, L., Bevilacqua, M., Milazzo, L., Cusi, D., Barlassina, C., Sarzi-Puttini, P., & Turiel, M. (2010). From endothelial dysfunction to atherosclerosis. Autoimmunity Reviews, 9(12), 830–834. https://doi.org/10.1016/j.autrev.2010.07.016
Fernando, S., Bursill, C. A., Nicholls, S. J., & Psaltis, P. J. (2020). Pathophysiology of Atherosclerosis. Mechanisms of Vascular Disease, 19–45. https://doi.org/10.1007/978-3-030-43683-4_2
Loffredo, L., & Carnevale, R. (2024). Oxidative Stress: The Hidden Catalyst Fueling Atherosclerosis and Cardiovascular Disease. Antioxidants, 13(9), 1089–1089. https://doi.org/10.3390/antiox13091089
Tasouli-Drakou, V., Ogurek, I., Shaikh, T., Ringor, M., DiCaro, M. V., & Lei, K. (2025). Atherosclerosis: A Comprehensive Review of Molecular Factors and Mechanisms. International Journal of Molecular Sciences, 26(3), 1364–1364. https://doi.org/10.3390/ijms26031364
Page, M. J., McKenzie, J. E., Bossuyt, P. M., Boutron, I., Hoffmann, T. C., Mulrow, C. D., Shamseer, L., Tetzlaff, J. M., Akl, E. A., Brennan, S. E., Chou, R., Glanville, J., Grimshaw, J. M., Hróbjartsson, A., Lalu, M. M., Li, T., Loder, E. W., Mayo-Wilson, E., McDonald, S., & McGuinness, L. A. (2021). The PRISMA 2020 statement: an Updated Guideline for Reporting Systematic Reviews. International Journal of Surgery, 88(105906), 105906. https://doi.org/10.1016/j.ijsu.2021.105906
Hung, K.-C., Kao, C.-L., Ho, C.-N., Wu, J.-Y., Chang, Y.-J., Lin, C.-M., & Chen, I-Wen. (2025). Efficacy and safety of esketamine in preventing perioperative neurocognitive disorders: a meta-analysis of randomized controlled studies. Systematic Reviews, 14(1). https://doi.org/10.1186/s13643-025-02807-1
Pepin, M. E., & Gupta, R. M. (2023). The role of endothelial cells in atherosclerosis: Insights from genetic association studies. American Journal of Pathology, 194(4). https://doi.org/10.1016/j.ajpath.2023.09.012
Rehberger Likozar, A., Zavrtanik, M., & Šebeštjen, M. (2020). Lipoprotein(a) in atherosclerosis: from pathophysiology to clinical relevance and treatment options. Annals of Medicine, 52(5), 162–177. https://doi.org/10.1080/07853890.2020.1775287
Linton, M. F., Yancey, P. G., Davies, S. S., Jerome, W. G., Linton, E. F., Song, W. L., Doran, A. C., & Vickers, K. C. (2019). The Role of Lipids and Lipoproteins in Atherosclerosis. Nih.gov; MDText.com, Inc. https://www.ncbi.nlm.nih.gov/books/NBK343489/
Amir Ajoolabady, Pratico, D., Lin, L., Mantzoros, C. S., Suhad Bahijri, Jaakko Tuomilehto, & Ren, J. (2024). Inflammation in atherosclerosis: pathophysiology and mechanisms. Cell Death and Disease, 15(11). https://doi.org/10.1038/s41419-024-07166-8
Poznyak, A. V., Nikiforov, N. G., Markin, A. M., Kashirskikh, D. A., Myasoedova, V. A., Gerasimova, E. V., & Orekhov, A. N. (2021). Overview of OxLDL and Its Impact on Cardiovascular Health: Focus on Atherosclerosis. Frontiers in Pharmacology, 11. https://doi.org/10.3389/fphar.2020.613780
Gui, Y., Zheng, H., & Cao, R. Y. (2022). Foam Cells in Atherosclerosis: Novel Insights Into Its Origins, Consequences, and Molecular Mechanisms. Frontiers in Cardiovascular Medicine, 9, 845942. https://doi.org/10.3389/fcvm.2022.845942
Amir Ajoolabady, Pratico, D., Lin, L., Mantzoros, C. S., Suhad Bahijri, Jaakko Tuomilehto, & Ren, J. (2024). Inflammation in atherosclerosis: pathophysiology and mechanisms. Cell Death and Disease, 15(11). https://doi.org/10.1038/s41419-024-07166-8
Gui, Y., Zheng, H., & Cao, R. Y. (2022). Foam Cells in Atherosclerosis: Novel Insights Into Its Origins, Consequences, and Molecular Mechanisms. Frontiers in Cardiovascular Medicine, 9, 845942. https://doi.org/10.3389/fcvm.2022.845942
Ouimet, M., Barrett, T. J., & Fisher, E. A. (2019). HDL and Reverse Cholesterol Transport. Circulation Research, 124(10), 1505–1518. https://doi.org/10.1161/circresaha.119.312617
Gui, Y., Zheng, H., & Cao, R. Y. (2022). Foam Cells in Atherosclerosis: Novel Insights Into Its Origins, Consequences, and Molecular Mechanisms. Frontiers in Cardiovascular Medicine, 9, 845942. https://doi.org/10.3389/fcvm.2022.845942
Ouimet, M., Barrett, T. J., & Fisher, E. A. (2019). HDL and Reverse Cholesterol Transport. Circulation Research, 124(10), 1505–1518. https://doi.org/10.1161/circresaha.119.312617
Filippatos, T. D., Kei, A., Rizos, C. V., & Elisaf, M. S. (2017). Effects of PCSK9 Inhibitors on Other than Low-Density Lipoprotein Cholesterol Lipid Variables. Journal of Cardiovascular Pharmacology and Therapeutics, 23(1), 3–12. https://doi.org/10.1177/1074248417724868
Sabatine, M. S., Giugliano, R. P., Keech, A. C., Honarpour, N., Wiviott, S. D., Murphy, S. A., Kuder, J. F., Wang, H., Liu, T., Wasserman, S. M., Sever, P. S., & Pedersen, T. R. (2017). Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. New England Journal of Medicine, 376(18), 1713–1722. https://doi.org/10.1056/nejmoa1615664
Gaudet, D., Watts, G. F., Robinson, J. G., Minini, P., Sasiela, W. J., Edelberg, J., Louie, M. J., & Raal, F. J. (2017). Effect of Alirocumab on Lipoprotein(a) Over ≥1.5 Years (from the Phase 3 ODYSSEY Program). The American Journal of Cardiology, 119(1), 40–46. https://doi.org/10.1016/j.amjcard.2016.09.010
Staels, B., Handelsman, Y., & Fonseca, V. (2010). Bile Acid Sequestrants for Lipid and Glucose Control. Current Diabetes Reports, 10(1), 70–77. https://doi.org/10.1007/s11892-009-0087-5
Zhang, B., Kuipers, F., de Boer, J. F., & Kuivenhoven, J. A. (2022). Modulation of Bile Acid Metabolism to Improve Plasma Lipid and Lipoprotein Profiles. Journal of Clinical Medicine, 11(1), 4. https://doi.org/10.3390/jcm11010004
Staels, B., Handelsman, Y., & Fonseca, V. (2010). Bile Acid Sequestrants for Lipid and Glucose Control. Current Diabetes Reports, 10(1), 70–77. https://doi.org/10.1007/s11892-009-0087-5
Kastelein, J. J. P., Akdim, F., Stroes, E. S. G., Zwinderman, A. H., Bots, M. L., Stalenhoef, A. F. H., Visseren, F. L. J., Sijbrands, E. J. G., Trip, M. D., Stein, E. A., Gaudet, D., Duivenvoorden, R., Veltri, E. P., Marais, A. D., de Groot, E., & ENHANCE Investigators. (2008). Simvastatin with or without ezetimibe in familial hypercholesterolemia. The New England Journal of Medicine, 358(14), 1431–1443. https://doi.org/10.1056/NEJMoa0800742
A Azezli, T Bayraktaroglu, F Kutluturk, A Cikim, S Tanyolac, & Y Orhan. (2006). Effects of Ezetimibe on serum lipoproteins in patients with homozygous familial hypercholesterolemia undergoing LDL cholesterol apheresis and high dose atorvastatin therapy. Endocrine Abstracts, 11. https://www.endocrine-abstracts.org/ea/0011/abstracts/poster-presentations/diabetes-metabolism-and-cardiovascular/ea0011p319/